آنسفالوپاتی
Die Enzephalopathie ( griechisch ἐγεφαλος enképhalos ، “مغز” آلمانی ، و یونان باستان πάθεια pátheia ، آلمانی “رنج” ) یک اصطلاح جمعی برای وضعیت های پاتولوژیک مغز با علل و ویژگی های مختلف است. نیز می نامیدند گاهی اوقات، روان پریشی ها را آنسفالوپاتی ، و در اینجا به طور خاص به روان پریشی با یا بدون کما در مورد نارسایی آدرنال به عنوان انسفالوپاتی اضافه می شود . [1]
ساختاری از آنجایی که ضایعات در بسیاری از موارد رخ نمی دهد، برگشت پذیری اغلب امکان پذیر است، اما همیشه داده نمی شود. این اصطلاح عموماً فقط برای تغییراتی استفاده میشود که بر کل مغز تأثیر میگذارد و نه فقط بر بخشهای مغز. ایجاد علائم بر اساس اختلال در عملکرد سلول های عصبی (نرون ها) و سلول های گلیال است . آنها به دلیل تغییر در محیط داخلی ارگانیسم و اختلال در هموستاز مغزی ایجاد می شوند که منجر به اختلال در عملکرد انتقال دهنده های عصبی و غشاء می شود . [2]
آسیب های طولانی مدت مانند اختلالات شناختی پس از آنسفالوپاتی ها احتمالاً می تواند به فرآیندهای التهابی عصبی ثانویه نیز مرتبط باشد، به عنوان مثال پس از سپسیس ، سوختگی یا عمل های طولانی مدت .
فهرست مطالب
طبقه بندی و علل
- آنسفالیت گیرنده ضد NMDA ، آنتی بادی علیه گیرنده NMDA
- انسفالوپاتی تروماتیک مزمن ، پس از ضربات یا برآمدگی های مکرر، همچنین پس از صرع
- انسفالوپاتی گلایسین ، ناشی از تولید بیش از حد گلیسین
- انسفالوپاتی کبدی ، در بیماری کبد، که احتمالاً توسط مواد آسیبرسان به مغز مانند آمونیاک ایجاد میشود .
- انسفالوپاتی هاشیموتو ، در تیروئیدیت خودایمنی
- انسفالوپاتی HIV در برخی از بیماران HIV دیده می شود
- انسفالوپاتی فشار خون بالا ( انسفالوپاتی هیپرتنسیوا [3] )، به دلیل فشار خون بالا
- هیپرآمونمی ، ناشی از سطوح بالای آمونیوم در خون
- هیپوکسی مغزی ، به دلیل کمبود اکسیژن
- انسفالوپاتی لایم ، در مراحل مزمن بیماری لایم .
- انسفالوپاتی میتوکندری ، ناشی از اختلال در DNA میتوکندری
- انسفالوپاتی شریانی اسکلروتیک زیر قشری (بیماری بینسوانگر)، ناشی از تصلب شرایین
- آنسفالوپاتی های اسفنجی شکل قابل انتقال (آنسفالوپاتی اسفنجی قابل انتقال، به اختصار TSE)، احتمالاً توسط پریون هایی مانند انسفالوپاتی اسفنجی شکل گاوی (BSE)، همچنین بیماری کروتسفلد-جاکوب ایجاد می شود.
- اورمیک انسفالوپاتی ، در نارسایی کلیوی
- ” آنسفالوپاتی Wernicke “، به دلیل کمبود ویتامین B1 ( تیامین )
انسفالوپاتی ها همچنین می توانند از جمله به دلیل غلظت غیر طبیعی مواد سمی ، برخی داروها [4] و الکترولیت ها ، توسط پاتوژن ها یا اختلالات گردش خون ایجاد شوند .
) نقش دارند داروها و داروها نیز از نظر تأثیرات سمی بر مغز ( توکسیدروم .
مسمومیت هایی مانند مسمومیت با سرب نیز می تواند علت انسفالوپاتی باشد. [5]
یک آنسفالوپاتی تروماتیک با منشاء مزمن، آنسفالوپاتی تروماتیک مزمن (CTE) است که در اصل و گاهی اوقات هنوز دمانس پوگیلیستیکا ([= زوال عقل بوکسور ]، انسفالوپاتی تروماتیکا بوکسورها ، انسفالوپاتی بوکسور [6] ) نامیده می شود . [7]
تظاهرات بالینی
علائم انسفالوپاتی تحت تسلط عصب روانپزشکی به علت خاص نیست و متنوع است: اختلالات هوشیاری ، اختلالات حرکتی ، دیستونی رویشی اغلب به عنوان بخشی از مشخصه به اصطلاح سندرم مغزی عمومی رخ می دهد . [8] کمتر رایج هستند علائم کانونی مغزی و علائم ساقه مغز ، به عنوان مثال. B. در هیپوگلیسمی یا انسفالوپاتی Wernicke. [9]
تشخیص
به عنوان یک آزمایش برای انسفالوپاتی ها، معاینه بالینی در مقایسه با الکتروانسفالوگرافی ( EEG)، با روش های نور رادیولوژیک و با تشخیص آزمایشگاهی مربوطه ( سرم و مشروب ) استفاده می شود. [2] تعیین دقیق سایر حاد و مزمن CNS اختلالات ، از جمله سکته مغزی ، عفونت، تروما، و صرع ، قبل از تشخیص انسفالوپاتی ضروری است.
در موارد انفرادی مانند بیماری مشکوک به پریون، از تکنیک تقویت چرخه ای تاشو اشتباه پروتئین (PMCA) استفاده می شود.
آنسفالوپاتی اسفنجی شکل قابل انتقال
| طبقه بندی بر اساس ICD-10 | |
|---|---|
| G81.9 [1] | عفونت ویروسی آتیپیک سیستم عصبی مرکزی، نامشخص |
| https://www.dimdi.de/static/de/klassifikationen/icd/icd-10-gm/kode-suche/htmlgm2023/block-g90-g99.htm | |
انسفالوپاتی اسفنجی قابل انتقال (TSE) یا انسفالوپاتی اسفنجی قابل انتقال نیز نامیده می شود که بیماری پریون در انسان ، مربوط به شکل قابل انتقال بیماری کروتزفلد-ژاکوب است و یکی از بیماری های تاخوردگی اشتباه پروتئین است . این یک بیماری مغزی به سرعت در حال پیشرفت ( انسفالوپاتی ) است که در آن یک تغییر اسفنجی در بافت مغز، از بین رفتن سلول های عصبی و گلیوز بدون علائم التهاب وجود دارد. بیماری ها هم در انسان و هم در حیوانات دیده می شود. علت آن هستند فرض بر این است که پریون ها نامیده می شود (از این رو بیماری پریون ). TSE ها همیشه کشنده هستند و در حال حاضر هیچ گزینه درمانی وجود ندارد.
فهرست مطالب
اشکال TSE
- قدیمی ترین TSE شناخته شده اسکراپی است ، یک بیماری اسفنجی مغز که در گوسفند و بز مشاهده می شود. در سال 1732 در انگلستان کشف شد.
- انسفالوپاتی اسفنجی شکل گاوی (BSE): در گاو، اولین بار در اواخر سال 1984 در انگلستان شناسایی شد.
- بیماری زوال مزمن (CWD): در گوزن و دیگر گوزن های آمریکای شمالی
- انسفالوپاتی ونگولات عجیب و غریب (EUE): در نایلاها و کودوس بزرگ (گونه های آنتلوپ آفریقایی)
- انسفالوپاتی اسفنجی شکل گربه (FSE): در گربه ها ، از سال 1990 تشخیص داده شده است.
- انسفالوپاتی راسو قابل انتقال (TME): در راسو ، اولین بار در سال 1947 در مزرعه راسو در ایالات متحده مشاهده شد.
گونههای حیوانی زیر نیز میتوانند تحت تأثیر TSE قرار گیرند: قاطر ، گوزن ، آهو، اوریکس، بابونه، موش صحرایی، خوکچه هندی، همستر، موش (در آزمایشهای آزمایشگاهی).
اتیولوژی
این بیماری ها به احتمال زیاد توسط پریون ها ایجاد می شوند .
آسیب شناسی
هیچ نشانه ای از پاسخ التهابی یا ایمنی در مغز بیمار وجود ندارد. ویژگی های مشخصه رسوبات رشته ای و پروتئینی در بافت عصبی و ساختار اسفنجی و سوراخ شده مغز است.
وظیفه
آنسفالوپاتی های اسفنجی شکل قابل انتقال باید در اتریش مطابق با بخش 1 بند 1 شماره 1 قانون اپیدمی 1950 در صورت سوء ظن، بیماری و مرگ گزارش شود . پزشکان و آزمایشگاهها، در میان دیگران، موظف به گزارش ( § 3 قانون اپیدمیها) هستند.
در آلمان، آنسفالوپاتی اسفنجی شکل انسانی (به استثنای اشکال خانوادگی-ارثی) باید طبق بخش 6 قانون حفاظت از عفونت (IfSG) در صورت مشکوک شدن، بیماری و مرگ از طرف پزشک و با نام گزارش شود. غیره دایره کسانی که موظف به گزارش هستند بر اساس بخش 8 IfSG است که طبق بخش 9 IfSG باید گزارش شود.
اهمیت اجتماعی
در سالهای 2000 تا 2005، گزارشهای مربوط به TSE در صفحه اول روزنامهها و در اخبار اصلی تلویزیون آلمان و سایر کشورها قرار داشت. [2] در اروپا، آگاهی از TSE منجر به تغییر میان مدت در مصرف گوشت گاو و کاهش قابل توجه (موقت) قیمت ها شد، در نتیجه بسیاری از شرکت ها مجبور شدند تولید خود را در کوتاه مدت تغییر دهند. قانونگذار . بسیاری از ایالت ها و اتحادیه اروپا قوانینی را صادر کردند که محصولات حیوانی باید آن را “ایمن” کنند (مثلاً مقررات BSE در آلمان) این نه تنها به محصولات گوشتی برای تغذیه و به عنوان خوراک دام، بلکه به عنوان مثال، به ژلاتین به عنوان ماده کپسول در داروها و حتی تسمه های چرمی برای اهداف ارتوپدی در دستگاه های پزشکی نیز گسترش می یابد . در سال 2001 آغاز و تأمین مالی شد در آلمان، تحقیقات پریون . [3]
در کره جنوبی در سال 2008 شورش های دسته جمعی در ارتباط با تسهیل واردات گوشت از ایالات متحده رخ داد که به پیامدهای سیاسی نیز منجر شد. [4]
بیماری کروتسفلد جاکوب
| طبقه بندی بر اساس ICD-10 | |
|---|---|
| A81.- | عفونت های ویروسی غیر معمول سیستم عصبی مرکزی |
| A81.0 | بیماری کروتسفلد جاکوب |
| F02.1* | زوال عقل در بیماری کروتسفلد-جاکوب |
| https://www.dimdi.de/static/de/klassifikationen/icd/icd-10-gm/kode-suche/htmlgm2023/block-g90-g99.htm | |
کروتسفلد بیماری CJD قابل انتقال جاکوب ( ) یک بیماری بسیار نادر و کشنده در انسان است که با پروتئین های غیر معمول (به اصطلاح پریون) مشخص می شود و یک انسفالوپاتی اسفنجی شکل ( مرتبط با انحلال اسفنجی بافت مغز) است . این بیماری نورودژنراتیو در انسان به صورت انتقالی، ژنتیکی یا پراکنده رخ می دهد. مشخصه این بیماری این است که پروتئینهای پریونی که بهطور غیرعادی چین خوردهاند، بهویژه در مغز، ساختار تغییر یافتهشان را به «عموزادههای» با ساختار سالمی که معمولاً در آنجا وجود دارند، تحمیل میکنند و بنابراین یک فرآیند بیوشیمیایی کشنده را در آنجا آغاز میکنند که در نهایت منجر به انحطاط میشود مغز _ . . پروتئین های چین خورده غیرطبیعی در سلول های عصبی رسوب کرده و توده هایی را تشکیل می دهند. عملکرد سلول های عصبی به طور فزاینده ای مختل می شود، به طوری که مرگ برنامه ریزی شده سلولی رخ می دهد ( آپوپتوز ). با پیشرفت بیماری، مغز آسیب دیده ساختاری اسفنجی و سوراخ دار با رسوبات رشته ای و پروتئینی به خود می گیرد. با این حال، در خون یک فرد بیمار، تنها مقدار کمی از پریون های عفونی وجود دارد
این بیماری در ابتدای دهه 1920 به نام کسانی که برای اولین بار آن را توصیف کردند، عصب شناس هانس گرهارد کروتسفلد (در برسلاو ) و (به طور مستقل) آلفونس ماریا یاکوب (در هامبورگ ) نامگذاری شد.
در سال 2017 موسسه روبرت کخ آلمان 77 بیماری و 78 مورد در سال 2018 گزارش کرده است. [1]
توضیحات اول
انحلال اسفنجی (اسفنجی شکل) بافت مغز در گوسفندان مبتلا به اسکرپی در سال 1732 در بریتانیا ثبت شد. [2]
معالجه کرد.او متخصص مغز و اعصاب در طول دوره آموزشی بیشتر در سال 1913، هانس گرهارد کروتسفلد، را در کلینیک روانپزشکی دانشگاه برسلاو تحت هدایت آلویس آلزایمر آینده از کیل، یک زن جوان مبتلا به اختلالات گفتاری ، گیجی و انقباضات عضلانی اندکی پس از آن درگذشت. به دلیل جنگ جهانی اول، او فقط توانست این “بیماری خاص و کانونی سیستم عصبی مرکزی” را در سال 1920 منتشر کند [3] ، اندکی قبل از آلفونس ماریا یاکوب ، متخصص مغز و اعصاب هامبورگ . [4]
این نام به نوروپاتولوژیست آلمانی والتر اسپیل مایر برمی گردد که در سال 1922 اصطلاح بیماری کروتسفلد-ژاکوب را پیشنهاد کرد. [5]
CJK/CJD
(TSE) است که در انسان یافت می شود این بیماری شایع ترین آنسفالوپاتی اسفنجی شکل قابل انتقال . CJK کلاسیک به سه شکل از قبل شناخته شده تقسیم می شود:
بیماری پریون پریون (sCJD)
شکل پراکنده بیماری کروتسفلد-جاکوب شایع ترین شکل بیماری در انسان در سراسر جهان است. عوامل محرک احتمالاً پریون ها هستند .
فرکانس
این عارضه در سرتاسر جهان با نرخ مشابه تقریباً 1 مورد در سال به ازای هر میلیون جمعیت رخ میدهد، اما اغلب در ابتدا اشتباه تشخیص داده میشود ، که بیشتر بهعنوان آنسفالیت ویروسی ، آتروفی مخچه پارانئوپلاستیک ، افسردگی ، سرگیجه یا بیماری آلزایمر است . [6] در آلمان، نسبت جنسیتی زنان به مردان 2:1 است. خطر ابتلا به این بیماری با افزایش سن افزایش می یابد و اوج بروز آن در حدود 70 سالگی است. در این سن، احتمال ابتلای سالانه به این بیماری حدود 1:125000 است. پس از آن، خطر دوباره کاهش می یابد. گاهی اوقات افراد جوان تر نیز بیمار می شوند. در آلمان، هر سال حدود 7 نفر زیر 50 سال به CJD پراکنده مبتلا می شوند. حتی جوانان نیز ممکن است بیمار شوند، اگرچه تنها تعداد انگشت شماری از این موارد در سراسر جهان شرح داده شده است. احتمال بیمار شدن قبل از 30 سالگی حدود 1:3,000,000 است.
سیر بیماری/علائم
این بیماری ابتدا به صورت موذیانه شروع می شود، اما بیمار مهارت های ذهنی و حرکتی خود را به طور اجتناب ناپذیر و سریع از دست می دهد. علائم زیر را می توان مشاهده کرد: ترسیدن، اختلالات حرکتی ( میوکلونوس ، آتاکسی )، اختلالات حافظه، اختلالات ادراک ( توهم ) و هوشیاری ، اختلالات بینایی و تغییرات شخصیتی، اختلالات رویشی و گیجی تا زوال عقل . مرحله آخر بیماری با mutism akinetic مشخص می شود . این بیماری معمولاً در عرض چند ماه منجر به مرگ می شود. این بیماری می تواند از 3-6 هفته تا بیش از 2 سال طول بکشد. میانگین طول مدت بیماری 4 تا 6 ماه است.
در شکل براونل-اپنهایمر ، که به عنوان نوع مخچه ( مخچه ) نیز شناخته می شود ، تنها نشانه هایی از اختلال مخچه بدون اختلالات شناختی در ماه اول بیماری وجود دارد. حدود 20 درصد از بیماری های پراکنده متعلق به این شکل است. معمولاً ابتدا ظاهر می شود با میانگین سن شروع 63 سال، راه رفتن ناپایدار ، سرگیجه و اختلالات هماهنگی ، اما پس از میانگین سه ماه، شکل کامل پیش رونده، از جمله اختلالات شناختی، ایجاد می شود. با این وجود، حتی با وجود شکل مخچه در MRI در توالیهای FLAIR منتشر و T2، معمولاً هیچ افزایش شدیدی در مخچه یافت نمیشود، اما در عقدههای قاعدهای و در تالاموس . [7]
بیماری پریون ژنتیکی
در این فرم، یک گروه کامل از بیماری های قابل ارثی در خانواده ها خلاصه می شود. همه این اشکال یک جهش خاص را به ارث می برند که منجر به یک پروتئین پریون معیوب می شود . این گروه از بیماری ها بسیار ناسازگار ( ناهمگن ) هستند و با علائم بالینی بسیار متغیر مشخص می شوند. اوج بیماری در اینجا حدود 50 سالگی و در نتیجه زودتر از شکل پراکنده است. طول مدت بیماری اغلب طولانی تر است. این اختلالات عبارتند از بیماری کروتسفلد-ژاکوب خانوادگی/ژنتیکی، سندرم گرستمن-اشتراوسلر-شاینکر (GSS) و بی خوابی فامیلی کشنده (FFI). شکل خانوادگی بیماری کروتسفلد-یاکوب توسط فردریش مگندوفر در اوایل سال 1930 توصیف شد. [8] [9]
شکل های انتقال یافته
انتقال مستقیم عامل بیماری زا از فردی به فرد دیگر تنها از طریق تماس با بافت عفونی به صورت ایتروژن (ناشی از پزشکان) ثابت شده است. مننژ و این اتفاق به ویژه در پیوندهای قرنیه و ابزارهای جراحی مغز و اعصاب که به اندازه کافی استریل نشده بودند، زودتر اتفاق افتاد. استخراج شده از هیپوفیز جسد مشاهده شده است انتقال مستقیم نیز با هورمون های رشد و گنادوتروپین های . در مجموع 132 مورد عفونت با هورمون رشد در سراسر جهان گزارش شده است که بیشتر این موارد از فرانسه و بریتانیا گزارش شده است. تاکنون هیچ موردی در آلمان گزارش نشده است، اگرچه بیمارانی با قد کوچک نیز در آنجا با هورمون های رشد درمان شده اند.
بیشتر موارد با پیوند مننژ مرتبط در ژاپن رخ داده است. این بیماری ها تقریباً منحصراً به محصول آلمانی Lyodura از B. Braun Melsungen AG نسبت داده می شود . این محصول به دلیل عدم کنترل بر روی اهداکنندگان مننژ و فرآیند تولید، که در آن مننژها به اندازه کافی ضدعفونی نشده و روی هم چیده شده بودند، که منجر به آلودگی متقاطع مننژهای سالم به پریونها میشد، خطرناک تلقی میشد. لیودورا به عنوان نوعی “گچ” نه تنها برای بازسازی مننژها، بلکه در انواع عمل های غیر جراحی مغز و اعصاب استفاده می شد، به خصوص که با واکنش های رد کم مشخص می شد. لیودورا باید در سال 1996 حذف می شد.
نوع جدید بیماری کروتسفلد-ژاکوب
در بریتانیا، در 20 مارس 1996 اعلام شد که چندین جوان بر اثر نوع جدیدی از CJD (nvCJD) جان خود را از دست داده اند.
انتقال

با توجه به دانش فعلی، احتمال 99٪ وجود دارد که این نوع (که اکنون به عنوان nvCJD، nv = نوع جدید = نوع جدید شناخته می شود) از خوردن گوشت گاو آلوده به BSE ناشی است. با این حال، احتمالاً اکثریت جمعیت به عفونت ناشی از مواد غذایی آلوده به BSE مقاوم هستند، زیرا تمام مبتلایان قبلی به nvCJD استعداد ژنتیکی داشتند که در کمتر از 40 درصد از جمعیت اروپا یافت می شود. [10] در یک نقطه بحرانی در ژنی که پروتئین پریون را کد می کند، آنها فقط دستورالعمل ترکیب اسید آمینه متیونین را پیدا کردند . با این حال، اکثریت جمعیت ناهمگن هستند و همچنین دارای ژنی هستند که باعث ادغام والین در این نقطه می شود. منطقی است که نتیجه بگیریم که پریون های انسانی در صورتی که حاوی آمینو اسید متیونین در محل تعیین شده باشند، به راحتی توسط پریون های BSE تا می شوند.

برخلاف بیماری پراکنده (sCJD)، که در خود مغز منشأ میگیرد و اساساً به سیستم عصبی مرکزی محدود میشود (مقدار کمی از پروتئین پریون غیرطبیعی نیز در اعصاب و ماهیچهها شناسایی شده است)، نوع جدید CJD (nvCJD) نیز تأثیر میگذارد. سیستم به اصطلاح لنفورتیکولار با غدد لنفاوی، طحال و لوزه ها (لوزه ها). بنابراین بیوپسی لوزه، برداشتن نمونه از لوزه های پالاتین با جراحی، می تواند برای تشخیص NVCJD استفاده شود. لوزه ها همیشه در nvCJD مثبت هستند در حالی که همیشه در CJD پراکنده (sCJD)، CJD ایتروژنیک (انتقال از انسان به انسان از طریق پیوند دورال یا عصاره هیپوفیز) و اشکال ارثی منفی هستند .
انتقال از طریق انتقال خون به طور تجربی در مدل های حیوانی ثابت شده است. [11] در بریتانیا، پیگیری دریافتکنندگان خون موارد vCJD، سه مورد را شناسایی کرد که به این بیماری نیز مبتلا شدند. [12]
نتایج تحقیقات جدید در دانشگاه زوریخ این نتیجه را می دهد که انتقال از طریق ذرات معلق در هوا امکان پذیر است. اما در عمل، این شکل از عفونت فقط باید در هنگام کار با لاشه حیوانات بیمار در کشتارگاه ها و آزمایشگاه ها نقش داشته باشد. [13]
تشخیص
در آگوست 2005، ، متخصص مغز و اعصاب کلودیو سوتو و همکارانش از دانشگاه تگزاس (ایالات متحده آمریکا) اعلام کردند که BSE و نوع جدید بیماری کروتسفلد-جاکوب اکنون با آزمایش خون قابل تشخیص هستند. سالم پریونهای محققان از خاصیت پریونهای عفونی تغییر یافته غیرطبیعی برای تحمیل ساختار خود بر سایر استفاده کردند تا پریونهای عفونی را که فقط به تعداد بسیار اندک در خون افراد بیمار وجود دارد، با ضریب ده میلیون ضرب کنند و بنابراین آنها را به راحتی بسازند. قابل تشخیص در یک سری آزمایش با همستر، پریون های عفونی را می توان با قابلیت اطمینان 89 درصد و بدون آلارم کاذب شناسایی کرد. همچنین کار بر روی کاربرد برای تشخیص در انسان و نظارت بر اهدای خون در حال انجام است.
پروتئین تا شده اشتباه به هضم پروتئاز مقاوم است ، اما شکل طبیعی PrP اینطور نیست. بنابراین پروتئین های معمولی را می توان در تشخیص “هضم” کرد. و اگر بقایایی ظاهر شود، باید از شکل بیماریزای پروتئین باشند.
جدای از آن، پریونهای nvCJD را میتوان در بافت لوزه، طحال یا آپاندیس مدتها قبل از حمله به مغز شناسایی کرد. بنابراین می توان بیوپسی لوزه را برای تایید ظن nvCJD انجام داد.
تصویربرداری رزونانس مغناطیسی
برای تشخیص، تصویربرداری رزونانس مغناطیسی تست تصویربرداری انتخابی برای مشکوک به NVCJD بالینی است. در بیش از 90 درصد موارد nvCJD تایید شده از نظر آسیب شناسی عصبی، به اصطلاح علامت پولوینار قبلاً در مراحل اولیه بیماری (2 تا 10 ماه پس از شروع علائم) در تالاموس مشهود بود . در بیشتر موارد، این امر امکان تشخیص بدون نیاز به بررسی های بیشتر را فراهم می کند. [14]
EEG
الکتروانسفالوگرافی . در اکثر موارد کندی منتشر را نشان می دهد، اما ممکن است تا زمانی که علائم روانپزشکی یا عصبی ظاهر نشود، قابل توجه نباشد
مایع مغزی نخاعی
بررسی CSF در پارامترهای استاندارد قابل توجه نیست، تعداد سلول های طبیعی، پروتئین تام و گلوکز وجود دارد، تنها به ندرت یک اختلال مانع خفیف تا متوسط وجود دارد. تشخیص پروتئین 14-3-3 در CSF نشانگر قابل اعتماد و حساسی برای بیماری پراکنده کروتزفلد-جاکوب (sCJD) است. با این حال، در مورد nvCJD، این تنها در 50٪ مثبت است. با این حال، مشخص شده است که افزایش غلظت CSF tau یک نشانگر حساس برای nvCJD است. منفی 14-3-3 دارای ارزش اخباری منفی 63 درصد و تاو منفی دارای ارزش اخباری منفی 81 درصد است. اگر هر دو تست منفی باشند، ارزش اخباری منفی برای vCJD به 84٪ افزایش می یابد [15]
سیر بیماری و علائم
سن متوسط شروع 26 سال (محدوده 12-74 سال) است. در 14 ماهگی (6-39)، طول مدت بیماری به طور غیرعادی بیشتر از شکل پراکنده است. اغلب بیماران در طول دوره بیماری ابتدا توسط روانپزشک معاینه شدند. مراحل اولیه نوع کروتزفلد-جاکوب تحت سلطه علائم روانپزشکی است، اما علائم عصبی در 15٪ موارد مقدم بر علائم روانپزشکی است و در 22٪ موارد از زمان شروع با علائم روانپزشکی همراه است. ویژگی های رایج روانپزشکی اولیه شامل نارسایی ، عصبانیت ، از دست دادن علاقه، بی خوابی ، اضطراب و کناره گیری بود که در بسیاری از موارد منجر به تشخیص افسردگی شد، اما در تعداد کمی از موارد نیز ویژگی های روان پریشی مانند توهمات شنیداری یا بینایی و پارانوئید یا هذیان ایجاد شد. بسیاری از بیماران دچار آشفتگی یا پرخاشگری میشدند که گاهی منجر به مشکلاتی در مدیریت میشد، اما افکار خودکشی نادر بود (9٪ موارد) و در هیچ موردی سابقه ای مبنی بر خودآزاری عمدی وجود نداشت. هذیان های زودگذر در گزارش های قبلی به عنوان یک ویژگی روانپزشکی غیرمعمول توصیف شده اند، و در حالی که در برخی موارد به وضوح یک ویژگی هستند، در مقایسه با فراوانی سایر علائم روانپزشکی آشکارتر، نسبتاً غیر معمول بودند. [16] [17] احتمال یک اختلال عصبی زمینهای در بسیاری از موارد با ایجاد اختلالات شناختی، از جمله حافظه ضعیف، مشکل در تمرکز، عدم تمرکز، یا در اقلیت، سردرگمی آشکار افزایش یافته است. این ویژگی ها به طور متوسط بین 4 تا 7.5 ماه از شروع بالینی ایجاد شدند، اگرچه در اقلیت کوچکی از موارد در مراحل اولیه وجود داشتند. شایع ترین ویژگی عصبی اولیه درد غیر مرتبط با علائم حسی بود. این حالت مداوم و ناراحت کننده بود و اغلب بر اندام ها ، تنه یا صورت تأثیر می گذاشت . بین چهار تا شش ماهگی، ویژگیهای عصبی رایج شامل اختلالات راه رفتن، اغلب به شکل بیثباتی جزئی و دیس آرتری بود . پارستزی و بی حسی با توزیعی مشابه درد اغلب در میانگین 4 تا 6 ماه رخ داد و تقریباً نیمی از بیماران را تحت تأثیر قرار داد. [18] ترکیب یک اختلال روانپزشکی با ویژگی های عاطفی یا روان پریشی و درد مداوم، دیس آرتری، آتاکسی راه رفتن ، یا علائم حسی حداقل باید باعث ایجاد سوء ظن نسبت به نوع بیماری کروتزفلد-ژاکوب شود، به ویژه هنگامی که با شواهدی از اختلال شناختی همراه باشد. برخی از ویژگیهای عصبی مانند علائم حسی، بیثباتی راه رفتن و دیس آرتری ممکن است در اختلالات روانپزشکی یا بهعنوان عوارض جانبی داروهای روانگردان وجود داشته باشد ، اما تداوم این علائم و ایجاد علائم عصبی اضافی ممکن است نشاندهنده کروتزفلد-ژاکوب باشد. گونه.
ویژگیهای عصبی نوع کروتسفلد-جاکوب مانند علائم مخچه، حرکات غیرارادی ( میوکلونوس ، کریا یا دیستونی فوقانی )، علائم نورون حرکتی و علائم بینایی بسیار شایع هستند اما نسبتاً دیر در سیر بیماری ظاهر میشوند. علائم روانپزشکی که احتمال یک علت ارگانیک را نشان می دهد ، به عنوان مثال. ب. سرگیجه، توهم و اختلال در مراقبت از خود نیز دیر ظاهر می شود.
در مرحله آخر، بیماران این بیماری دیگر فرصتی برای برقراری ارتباط با محیط خود و یا واکنش به آن ندارند. به همین دلیل است که مبتلایان مرحله نهایی nvCJD اغلب به عنوان “مردگان زنده” شناخته می شوند. کامل گاهی اوقات این منجر به فلج اسپاستیک بدن می شود که اصطلاحاً به آن سفتی مغزی می گویند . بیماران برای مدت طولانی در این مرحله پایانی بیماری (وضعیت پایانی) باقی می مانند تا زمانی که بمیرند، به ویژه در اثر ذات الریه یا فلج تنفسی .
شیوع و فراوانی
تا سال 2014، 177 مورد در بریتانیا و 51 مورد در 11 کشور دیگر از نوع جدید CJD ثبت شده است. اکثر موارد در خارج از بریتانیا در فرانسه رخ داده است و به واردات گوشت گاو بریتانیا نسبت داده می شود. مرگ و میر در بریتانیا در سال 2000 به اوج خود رسید و از آن زمان تاکنون موارد بسیار نادری گزارش شده است. تعداد موارد در خارج از کشور در سال 2005 و 2006 به اوج خود رسید. بررسی 32441 آماده سازی آپاندیس برداشته شده وجود پریون PrPSc بالقوه بیماری زا را در 16 مورد نشان داد. در چندین مطالعه با مجموع حدود 44000 نمونه از لوزه های برداشته شده، یک نمونه مثبت پیدا شد. [19] تا مارس 2021، هیچ موردی از nvCJD در آلمان گزارش نشده است. [20]
درمان
از سال 2022، هیچ درمان و درمان موثری برای بیماری کروتسفلد-جاکوب وجود ندارد. تاکنون تنها درمان علامتی و تسکینی امکان پذیر است. [21] [22]
به عنوان بخشی از یک تلاش درمانی فردی، 6 بیمار بین سالهای 2018 تا 2022 در دانشگاه کالج لندن تحت درمان تجربی قرار گرفتند . این یک آنتی بادی مونوکلونال (نام توسعه PRN100) علیه پروتئین پریون (PrP) بود. این درمان بدون عوارض جانبی قابل توجهی به خوبی تحمل شد و آنتی بادی ها توانستند به مغز برسند، جایی که ممکن است به خنثی کردن پریون ها کمک کنند. با وجود این، بیماران همچنان وخامت وضعیت عصبی خود را نشان میدهند و به طور کلی بیش از حد انتظار با سیر طبیعی بیماری زنده نمیمانند. با این حال، نتایج مطالعه توسط محققان درگیر به عنوان امیدوارکننده تفسیر میشود، به همین دلیل است که آنها مطالعه بزرگتری را با شروع درمان در مرحلهای زودتر از سیر بیماری پیشنهاد میکنند. [23] [24]
بیماری تاخوردگی اشتباه پروتئین
به بیماریهای تاخوردگی نادرست پروتئین نیز بیماریهای تاخوردگی و خارج پروتئین تاخوردگی نادرست پروتئینها گفته میشود بیماریهایی ایجاد میشوند . ، هستند که که در اثر در داخل سلولها تجزیه می شوند پروتئین های تا شده اشتباه یا در سلول ها ذخیره می شوند یا در پروتئازوم . (پلاک) تشکیل می شود سمی در مورد اول، رسوبات ، در مورد دوم به دلیل کمبود پروتئین مربوطه در سلول یا در کل ارگانیسم ، عملکرد خود را از دست می دهد . هر دو می توانند در طول زمان برای افراد مبتلا آسیب شناسی شوند و بسته به پروتئین آسیب دیده منجر به بیماری های مختلفی شوند.
مکانیسم بیوشیمیایی
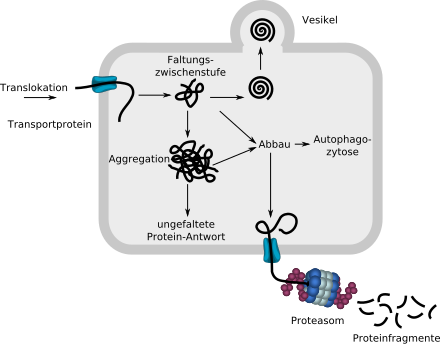
به ER وارد می شود پروتئین تا شده خطی که از طریق یک پروتئین حمل و نقل با کمک چاپرون شروع به تا شدن می کند (نمایش داده نشده است). اگر توسط کنترل کیفیت پروتئین به درستی تا شده تشخیص داده شود، از طریق یک وزیکول از ER خارج می شود. پروتئین های تا شده اشتباه از طریق یک پروتئین حمل و نقل به داخل سیتوزول قاچاق می شوند و در یک پروتئازوم به قطعات تقسیم می شوند. سنگدانه های آمورف نیز می توانند از طریق اتوفاژی تجزیه شوند .
اگر تعداد زیادی مولکول به دلیل تا شدن نادرست شکسته شوند، می تواند منجر به از دست دادن عملکرد در سلول یا کل ارگانیسم شود. اگر توده های نامحلول و غیر قابل تجزیه بیش از حد زیادی تشکیل شوند، رسوبات سمی برای سلول و کل ارگانیسم ایجاد می شود. [1]
در اکثر سلول های همه موجودات به عنوان بخشی از بیوسنتز پروتئین (پروتئین ها) به طور مداوم ، طیف گسترده ای از پروتئین ها تولید می شوند که عملکردهای گسترده ای را در سلول و در کل ارگانیسم انجام می دهند. برای عملکرد صحیح یک پروتئین، ساختار سوم آن از اهمیت حیاتی برخوردار است. این ساختار از طریق فرآیندی به نام تاخوردگی پروتئین به دست می آید. تا کردن پروتئین یک فرآیند پیچیده و ظریف است. تاخوردگی صحیح پروتئین توسط کنترل کیفیت پروتئین کنترل می شود . از نظر آماری، حدود 30 درصد از تمام پروتئین های حاصل از بیوسنتز پروتئین به اشتباه تا شده و به طور معمول در پروتئازوم سلول در حدود ده دقیقه تجزیه می شوند. [2] [3] انباشتگی پروتئینهای تاخورده نادرست در شبکه آندوپلاسمی منجر به پاسخ پروتئین بازشده با سرکوب ترجمه و افزایش سنتز چپرونها ، یک پاسخ استرس سلولی مرتبط میشود.
یک مولکول پروتئینی که به اشتباه تاخورده شده است، مسئول تصاویر بالینی جدی بیماری های تاخوردگی اشتباه پروتئین نیست. برای این کار باید مقادیر زیادی از این پروتئین ها تولید شود یا تعداد مولکول های به درستی تا شده کاهش یابد. در پریونها ، این اتفاق میافتد زیرا یک مولکول بهطور نادرست تا شده، وقتی با یک مولکول درست تا شده تماس پیدا میکند، باعث میشود مولکول صحیح باز شود و در نهایت به اشتباه تا شود. از آنجایی که اولین پروتئین اشتباه تا شده توسط این فرآیند تغییر نمی کند (بنابراین به عنوان یک آنزیم عمل می کند )، سپس دو مولکول به اشتباه تا شده وجود دارد. اینها می توانند مولکول های صحیح بیشتری را تا کنند.
به پروتئین های معیوب به عنوان محصولات معیوب ( DRiPs ریبوزومی ) نیز گفته می شود. [4]
علل
دلایل تاخوردگی نامناسب پروتئین پیچیده است. جهشهای ژنی در اگزونها که به تغییر در توالی اسید آمینه ، یعنی ساختار اولیه محصول ژن منجر میشود، تأثیر مستقیمی بر ساختار ثانویه و سوم یا بر روی سینتیک چینخوردگی پروتئین دارد. اشتباهات در رونویسی یا ترجمه نیز می تواند منجر به تا زدن اشتباه پروتئین شود. یکی دیگر از عوامل احتمالی محیط است. به عنوان مثال، در بیماری های عفونی پریون ، پروتئین پریون با غذا خورده می شود یا با ابزار جراحی منتقل می شود. در این میان، اولین نشانههای تولید شده توسط سیکادها و سیانوباکتریها سمی ( BMAA ) نیز وجود دارد که از طریق الحاق آن به پروتئینها، منجر به تا شدن نادرست آنها و در نتیجه احتمالاً شکلی از ALS میشود . [5]
بهرهگیری از عملکرد سمی
اگر DRiP های موجود در پروتئازوم نتوانند تجزیه شوند، مثلاً به دلیل اینکه قبلاً به صورت توده ها جمع شده اند، DRiP ها در سلول جمع می شوند. در آنجا آنها می توانند در طول زمان آسیب شناسی شوند، یعنی منجر به بیماری های خاص شوند. توده های پروتئین در درجه اول منجر به بیماری های تخریب کننده عصبی مانند بیماری پارکینسون ، بیماری آلزایمر یا بیماری هانتینگتون می شوند . دانه ها عملکرد سمی جدیدی در سلول ها دارند. اصطلاح افزایش عملکرد (سمی) برای اثر سمی درون سلول ها استفاده می شود. [6]
در ادبیات تخصصی انگلیسی زبان، اصطلاحات پروتئینوپاتی و پروتئوپاتی برای این شکل از بیماریهای تاخوردگی اشتباه پروتئین ایجاد شدهاند. از سوی دیگر، اصطلاحات آلمانی مربوط به پروتئوپاتی و پروتئینوپاتی ( پیشوند پروتئو- = ‘ پروتئین ‘ و پسوند -pathie = ‘بیماری’)، از سوی دیگر، به سختی در ادبیات تخصصی آلمانی زبان پیاده سازی شده اند.
در حال حاضر (از سال 2011) بیش از 100 پروتئینوپاتی در انسان و حیوان شناخته شده است. آنها در اثر رسوب حدود 20 پروتئین غیر همولوگ ایجاد می شوند. گروه بزرگ و مهمی را تشکیل می دهند آمیلوئیدوزها . [7]
بیماری های تاخوردگی اشتباه پروتئین با افزایش عملکرد سمی شامل بیماری های زیر است:
| بیماری | پروتئین ایجاد کننده | ملاحظات |
| بیماری آلزایمر [8] | β-آمیلوئید ، تاو | نوع تائوپاتی |
| بیماری پیک | آره | نوع تائوپاتی |
| دژنراسیون کورتیکوبازال | آره | نوع تائوپاتی |
| بیماری دانه نقره | آره | نوع تائوپاتی |
| فلج فوق هسته ای پیشرونده | آره | نوع تائوپاتی |
| بیماری پارکینسون [8] | α-سینوکلئین | نوع سینوکلئینوپاتی |
| مولتی سیستماتروفی | α-سینوکلئین | نوع سینوکلئینوپاتی |
| زوال عقل بدن لوی | α-سینوکلئین | نوع سینوکلئینوپاتی |
| کوریا هانتینگتون [8] | هانتینگتین | بیماری پلی گلوتامین گسترش از خانواده بیماری های تری نوکلئوتید |
| آتاکسی نخاعی مخچه ای [9] | تو آ. آتاکسین-2 | بیماری پلی گلوتامین |
| آتروفی عضلانی اسپینوبولبار نوع کندی [10] | آندروژنرزپتور | بیماری پلی گلوتامین |
| Dentatorubro-Pallidoluysische Atrophie (DRPLA) | آتروفین | بیماری پلی گلوتامین |
| آمیلوئیدوز ATTR و آمیلوئیدوز AP | ترانس تیرتین | آمیلوئیدوز |
| آمیلوئیدوز سیستمیک ارثی | (معمولا) ترانس تیرتین | آمیلوئیدوز |
| آمیلوئیدوز سیستمیک غیر ارثی [11] | زنجیره های سبک ایمونوگلوبولین (نوع AL)، APP قطعات (نوع AA)، β2-میکروگلوبولین (نوع AB) | آمیلوئیدوز |
| بیماری کروتسفلد جاکوب [12] | پریون ها | بیماری های پریون، از جمله همچنین بی خوابی فامیلی کشنده ، سندرم گرستمن-اشتراوسلر-شاینکر ، کورو |
| آب مروارید (آب مروارید) [13] | پروتئین های متعدد | دناتوره کردن مختلف عدسی پروتئین های |
| اسکلروز جانبی آمیوتروفیک (حداقل در برخی از انواع بیماری) [14] | TDP-43، FUS، SOD1 | |
| بیماری اسکندر [15] | پروتئین اسیدی فیبریلاری گلیال (GFAP) | |
| کادازیل [16] | شکاف 3 | |
| کم خونی داسی شکل | هموگلوبین | |
| Frontotemporallappen-Degeneration (FTLD-TLP) | TDP-43 | |
| آلوئولارپروتئینوز | سورفکتانت -پروتئین-C | |
| میوزیت بدن انکلوژن پراکنده [17] | بتا آمیلوئید (هنوز تایید نشده است) | |
| دیابت نوع 2 [18] | آمیلین |
از دست دادن عملکرد فیزیولوژیکی
بیماریهای تاخوردگی اشتباه پروتئین نیز شامل بیماریهایی میشوند که در آن پروتئینهای تاخورده اشتباه در پروتئازوم شکسته میشوند و در نتیجه مقادیر ناکافی پروتئین در دسترس سلولها یا ارگانیسم قرار میگیرد. [19] این از دست دادن عملکرد، engl. از دست دادن عملکرد (فیزیولوژیکی) می تواند منجر به بیماری هایی مانند فیبروز کیستیک شود . [20] اکثر بیماران مبتلا به فیبروز کیستیک دارای جهش ΔF508 ( نوع حذف ) در پروتئین CFTR ، یک کانال کلرید هستند . حذف سه نوکلئوتید باعث می شود که اسید آمینه فنیل آلانین (در کد یک حرفی F) در موقعیت 508 CFTR وجود نداشته باشد. این جهش سینتیک تاشدگی CFTR بسیار پیچیده را که دارای 21 گذرنده حوزه پروتئینی است ، به شدت تغییر می دهد. فرآیند تا شدن CFTR نوع وحشی در حال حاضر بیش از دو ساعت طول می کشد و تنها حدود 30 درصد از مولکول های CFTR سنتز شده به اندازه کافی سریع تا می شوند تا از تخریب پروتئین مرتبط با ER (ERAD) فرار کنند . ΔF508-CFTR حتی بدتر تا می شود و کاملاً تخریب می شود، اگرچه در اصل به عنوان یک کانال یونی کاملاً کاربردی است. بیماران مبتلا به این جهش فاقد کانال کلرید (= از دست دادن عملکرد) هستند، به این معنی که ترکیب ترشحات دفعی مختلف غدد به شدت تغییر می کند. [21] [22]
از بین رفتن عملکرد فیزیولوژیکی در بیماری های زیر وجود دارد، از جمله:
| بیماری | پروتئین/ ژن معیوب | ملاحظات |
| کلیه های کیستیک [23] | پلی سیستین-1 | |
| Morbus Charcot-Marie-Tooth [24] | آمینواسیل-tRNA-سنتتاز (AARS) | |
| سندرم لنفوپرولیفراتیو کروموزومی X [25] | SH2D1A | |
| موربوس هیرشپرونگ [26] | Rezeptor-Tyrosinkinase Ret | |
| هموسیستینوری و اسیدوری متیل مالونیک [27] | MMACHC | |
| Patellahypoplasie [28] | TBX4 | دیسپلازی ایسکیوپتلار |
| اسکلرواستئوس [29] | اسکلروستین | |
| CF [30] [31] | CFTR | |
| فنیل کتونوری [32] | فنیل آلانین هیدروکسیلاز | |
| سندرم دست-پا-تناسلی [33] | Homöobox-Protein A13 | |
| بیماری های ذخیره لیزوزومی [34] | آنزیم های مختلف لیزوزومی | بیش از 40 بیماری فردی، از جمله بیماری گوچر [35] ، بیماری فابری [36] سندرم تای ساکس [37] یا بیماری کراب [38] |
| سندرم QT [39] | تو آ. hERG | |
| سندرم آنجلمن | UBE3A | |
| ارثی سرطان سینه [40] | BRCA1 |
افزایش عملکرد و از دست دادن عملکرد
علاوه بر این، بیماریهای تاخوردگی نادرست پروتئین وجود دارد که در آنها هم از دست دادن عملکرد و هم رسوبات پروتئین سمی میتواند پاتولوژیک شود. نمونه ای از این کمبود آلفا-1 آنتی تریپسین است . یک جهش در ژن SERPINA1 که پروتئین فاز حاد α-1-آنتی تریپسین -یک مهارکننده پروتئاز- را ، رمزگذاری می کند باعث تا شدن نادرست α-1-آنتی تریپسین می شود. α-1-آنتی تریپسین عمدتاً توسط سلول های کبدی در کبد بیان می شود . به دلیل تا شدن نادرست، نمی تواند توسط هپتوسیت ها ترشح شود و رسوبات داخل سلولی را تشکیل می دهد. از دست دادن عملکرد منجر به آمفیزم پیشرونده ریه در بیماران مبتلا می شود ، زیرا کمبود α-1-آنتی تریپسین باعث می شود آنزیم لکوسیت الاستاز ( لوکوسیت الاستاز انسانی ، HLE) ساختار ریه را بدون کنترل تخریب کند. کبدی موازی با آمفیزم ریوی رسوب آلفا-1-آنتی تریپسین در سلول های کبدی منجر به سیروز می شود . [20] [41] [42]
مفاهیم درمانی

در حال حاضر بیماری های تا شدن اشتباه پروتئین قابل درمان نیستند. برای شایعترین بیماریهای نورودژنراتیو که در اثر افزایش عملکرد سمی ایجاد میشوند علّی یا درمانی هنوز هیچ درمان ، وجود ندارد. بیماران معمولاً به صورت علامتی یا صرفاً تسکین دهنده درمان می شوند . برخی از مفاهیم درمانی درمانی آینده مانند ژن درمانی وجود دارد که هنوز سال ها تا تایید آنها فاصله دارد .
بیماری های تاخوردگی نادرست پروتئین که به دلیل از دست دادن عملکرد پروتئین ایجاد می شوند، در برخی موارد قابل درمان هستند. در درمان جایگزینی آنزیم ، پروتئین از دست رفته که توسط مهندسی ژنتیک تولید می شود، می به طور مصنوعی به بدن بیمار تزریق شود. درمانهای چاپرون ممکن است گزینههای درمانی آینده برای هر دو نوع بیماری نادرست پروتئین باشد. [43] مولکولی چاپرون های پروتئین هایی هستند که نقش اصلی آنها “کمک به” تا شدن صحیح پروتئین های تازه سنتز شده است. علاوه بر این، شیمیایی و چاپرون های دارویی “مصنوعی” شناسایی و توسعه یافته اند که از فرآیند تا شدن نیز پشتیبانی می کنند. [44] داروی ساپروپترین، که برای درمان فنیل کتونوری استفاده میشود، نمونهای از داروساز تایید شده دارویی است. iminosugar . 1-deoxygalactonojirimycin (DGJ)، نام غیر انحصاری بین المللی migalastat ، یکی دیگر از عوامل دارویی است که در حال حاضر (از اکتبر 2011) در فاز III آزمایش بالینی برای اثربخشی در بیماران مبتلا به بیماری فابری است [45]
(EGCG) که عمدتاً در چای سبز یافت اپی گالوکاتچین گالات می شود ، ظاهراً می تواند از تا شدن صحیح پروتئین ها پشتیبانی کند. [46] در آزمایشگاهی آزمایشهای ، EGCG توانست فیبریلوژنز (تشکیل فیبریلها ) هانتینگتین، [47] α-سینوکلئین و β-آمیلوئید را مهار کند. [48] [49] [50] EGCG تضمین می کند که الیگومرهای کروی بی ضرر به جای فیبرهای سمی فیبری تشکیل می شوند. ظاهراً میتواند پلاکهایی را که قبلاً تشکیل شدهاند نیز حل کند. [48] در موش های رنگی، سطح پلاک در قشر، هیپوکامپ و قشر آنتورینال هر کدام حدود 50 درصد کاهش یافت. [51]
تا کردن اشتباه خارج از سلول ها
از حدود سال 2008، شناخت فزاینده ای وجود داشته است که تا کردن اشتباه پروتئین نه تنها منجر به مشکلاتی در داخل سلول ها می شود، بلکه تا حد قابل توجهی در فضای بین سلولی (بین سلولی) نیز منجر می شود. [52] اهمیت سیستم گلیمفاتیک (دفع مواد زائد سیستم عصبی مرکزی ) برای حذف پروتئینهای اشتباه تا شده از مغز در سال 2012 کشف شد و از آن زمان تاکنون موضوع تحقیقات فشرده بوده است. این امر به ویژه در مورد همه بیماری های عصبی شناخته شده و گسترده صدق می کند . [53]
گلیوز
| طبقه بندی بر اساس ICD-10 | |
|---|---|
| G93.8 [1] | سایر بیماری های مشخص شده مغز |
| H35.3 [1] | دژنراسیون ماکولا و قطب خلفی |
| https://www.dimdi.de/static/de/klassifikationen/icd/icd-10-gm/kode-suche/htmlgm2023/block-g90-g99.htm | |
گلیوز اصطلاحی برای افزایش تعداد سلول های گلیال در ناحیه آسیب دیده سیستم عصبی مرکزی یا مغز است . غیر اختصاصی این پاسخ آسیب شناسی عصبی به بیماری های عصبی مختلف است . سلول های گلیال فضا را اشغال می کنند اما عملکرد سلول های عصبی تخریب شده را نمی گیرند .
گلیوز اپی رتینال
گلیوز اپی رتینال ، همچنین پوکر ماکولا ، به دلیل محل آن در چشم بر خلاف گلیوز بقیه سیستم عصبی مرکزی، مستقیماً قابل مشاهده است. روی غشای مرزی داخلی (اپیتینال) بین شبکیه و جسم زجاجیه در این بیماری ، سلولهای کلاژندار انقباضی که میتوانند منشأ بسیار متفاوتی داشته باشند، جمع میشوند، جایی که لایهای متراکم را تشکیل میدهند که تمایل به کوچک شدن تدریجی دارد. سلولهای احتمالی منشا شامل آستروسیتها و سلولهای مولر شبکیه، یعنی سلولهای گلیال واقعی، اما همچنین سلولهای اپیتلیوم رنگدانه شبکیه یا سلولهای التهابی هستند.
علت
گلیوز اپی رتینال می تواند بدون علت ظاهری ( ایدیوپاتیک ) یا در نتیجه بیماری چشم یا جراحی (ثانویه) رخ دهد.
علائم
بسته به وسعت آن، گلیوز اپی رتینال می تواند مورد توجه قرار نگیرد اما همچنین می تواند باعث اختلالات بینایی مانند دگرگونی یا اسکوتوم مرکزی شود . غشاهای گلیوتیک بسیار گسترده و منقبض می توانند منجر به جدا شدن شبکیه و از دست دادن کامل بینایی در چشم آسیب دیده شوند.
تشخیص
در افتالموسکوپ ، غشای اپی رتینال را می توان با درخشش آن و هرگونه انحراف مشیمیه و عروقی که ممکن است رخ داده باشد، تشخیص داد. آزمون با شبکه Amsler برای تعیین دگرگونی اولیه و دینامیک آن استفاده می شود.
درمان
گلیوزهای مکرر و ظریف اپی رتینال بدون تمایل به پیشرفت نیازی به درمان ندارند.
در صورت بروز علائم مزاحم، بهبود بینایی یا در برخی موارد بهبودی کامل با کندن غشاهای اپی رتینال امکان پذیر است . عملی که در آن پس از ویترکتومی پارس پلانا تمام غشای محدود کننده اپی رتینال برداشته می شود.
بیماری هایی با علائم مشابه
علائم مشابهی می تواند توسط بسیاری از بیماری های چشمی دیگر به خصوص شبکیه و مشیمیه ایجاد شود و خود این بیماری می تواند نشانه ای از یک بیماری چشمی دیگر باشد، به طوری که چنین علائمی همیشه نیاز به معاینه کامل کل چشم آسیب دیده و طرف مقابل دارد.
گلیسین
| فرمول ساختاری | ||||
|---|---|---|---|---|
 |
||||
| عمومی | ||||
| نام | گلیسین | |||
| نامهای دیگر |
|
|||
| فرمول مولکولی | C2H5NO2 _ _ _ _ _ | |||
| توضیح کوتاه | جامد کریستالی بی رنگ و بی بو [3] | |||
| شناسه های خارجی / پایگاه های داده | ||||
|
||||
|
|
|||||||||
| اطلاعات دارویی | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ATC-Code | B05 CX03 | ||||||||
| مشخصات | |||||||||
| توده های مولی | 75,07 گرم · mol -1 | ||||||||
| حالت فیزیکی | جشن | ||||||||
| تراکم | 1,161 گرم در سانتی متر 3-3 [3] | ||||||||
| نقطه ذوب | تجزیه: 232-236 درجه سانتیگراد [3] | ||||||||
| p K S -Wert |
|
||||||||
| انحلال پذیری | به راحتی در آب حل می شود [6]
|
||||||||
| دستورالعمل های ایمنی | |||||||||
|
|||||||||
| داده های سم شناسی | 7930 mg·kg -1 ( LD 50 ، Ratte ، خوراکی ) [3] | ||||||||
| خواص ترمودینامیکی | |||||||||
| ΔH f 0 | -528.5 کیلوژول بر مول [7] | ||||||||
| تا جایی که ممکن و مرسوم باشد، از واحدهای SI استفاده می شود. مگر اینکه غیر از این ذکر شود، داده های داده شده تحت شرایط استاندارد اعمال می شود . | |||||||||
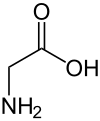
گلیسین ، به اختصار Gly یا G ، (همچنین گلیسین یا گلیکوکول ، از یونانی باستان κόλλα kólla: چسب، بر اساس نامگذاری شیمیایی سیستماتیک اسید آمینه استیک یا اسید آمینه اتانوئیک)، کوچکترین و سادهترین α اسید آمینه است و اولین بار در سال 1820 از ژلاتین ساخته شد. من. اچ. از هیدرولیز کلاژن مشتق شده این اسید آمینه به گروه اسیدهای آمینه آبدوست تعلق دارد و تنها پروتئین زا اسید آمینه (یا پروتئین ساز) است که غیر کایرال است و بنابراین از نظر نوری فعال نیست .
گلیسین ضروری نیست، بنابراین می تواند توسط خود ارگانیسم انسان سنتز شود و یک جزء مهم تقریباً همه پروتئین ها و یک گره مهم در متابولیسم است.
این نام از طعم شیرین گلیسین خالص ( یونانی γλυκύς glykýs ، آلمانی “شیرین” ).
تاریخچه
گلیسین اولین اسید آمینه ای است که از هضم اسیدی پروتئین ها به دست می آید. در نانسی در سال 1819 به دست آمد این امر توسط هانری براکونوت که با هدف استخراج قند از مواد حیوانی، چسب را با اسید سولفوریک هیدرولیز کرد.

بنابراین او کریستال های خوش طعمی که پس از تصفیه به دست می آید sucre de gélatine را به انگلیسی «شکر چسبان» نامید. [8] ژلاتین جزء اصلی چسب گلوتن است .
اندکی بعد، این ماده به گلیکوکول (“چسب شیرین”) تغییر نام داد، قبل از اینکه یونس یاکوب برزلیوس در سال 1848 تصمیم گرفت که از این پس از نام کوتاهتر گلیسین استفاده کند . ساختار شیمیایی تنها در سال 1858 توسط شیمیدان فرانسوی آگوست آندره توماس کاهورز به درستی توصیف شد . [9]
سنتز
) که در طی واکنش فرمالدئید ، هیدروژن سیانید و آمونیاک ( سنتز Strecker ) تشکیل می شود آمینو نیتریل (به طور دقیق تر: α- آمینو استونیتریل ، در هیدرولیز گلیسین تولید می کند:
- اچ سی اچ O + اچ سی ن + ن اچ 3 ⟶ اچ 2 ن – سی اچ 2 – سی ن ⟶ اچ 2 ن – سی اچ 2 – سی O O اچ



به عنوان یک واکنش جزئی، این واکنش نقش ویژهای در این فرضیه داشت که مولکولهای آلی بهعنوان «بلوکهای سازنده» برای اولین موجودات اولیه حدود 4 میلیارد سال پیش از ترکیبات معدنی ساده جو اولیه زمین پدید آمده بودند . ترکیبی از آب (H2O ) ، متان (CH4 ) ، آمونیاک (NH3 ) ، هیدروژن (H2 ) و مونوکسید کربن (CO) و همچنین هلیوم (He) و سایر گازهای نجیب برای این جو اولیه در نظر گرفته شده است. (به آزمایش میلر-اوری مراجعه کنید ).
از نظر شیمیایی، گلیسین همچنین می تواند از اسید مونوکلرواستیک و آمونیاک تولید شود:
- سی ل سی اچ 2 سی O O اچ + ن اچ 3 + ن آ O اچ ⟶ اچ 2 ن – سی اچ 2 – سی O O اچ + اچ 2 O + ن آ سی ل


در بدن، بیشتر گلیسین از غذا به دست می آید، اما می توان آن را از سرین نیز تهیه کرد .
خواص
وجود دارد گلیسین عمدتاً به عنوان یک “نمک درونی” یا زوئیتریون ، که تشکیل آن را می توان با این واقعیت توضیح داد که پروتون گروه اسیدی کربوکسی به جفت الکترون های تنها روی اتم نیتروژن گروه آمینه پایه مهاجرت می کند :

زوئیتریون در میدان الکتریکی مهاجرت نمی کند زیرا در کل بدون بار است. به بیان دقیق، این مورد در نقطه ایزوالکتریک (در یک مقدار pH مشخص، در اینجا 5.97 [10] ) است، که در آن گلیسین نیز کمترین انحلال خود را در آب دارد.
- Van-der-Waals-Volumen : 48
- درجه آبگریزی : -0.4
گلیسین آزاد طعم شیرینی دارد و آستانه تشخیص آن بین 25 تا 35 میلی مول در لیتر است . [11]
وقوع
مثالهای زیر نمای کلی از محتوای گلیسین را ارائه میدهند و هر کدام به 100 گرم غذا مربوط میشوند. درصد گلیسین نسبت به کل پروتئین نیز آورده شده است: [12 ]
| مواد غذایی | در مجموع- پروتئین |
گلیسین | بخش |
|---|---|---|---|
| گوشت خوک ، خام | 21 گرم | 0.95 گرم | 4.5 % |
| فیله سینه مرغ خام | 21 گرم | 0.95 گرم | 4.4 % |
| ماهی قزل آلا، خام | 20.5 گرم | 0.95 گرم | 4.7 % |
| پودر ژلاتین ، شیرین نشده | 86 گرم | 19 گرم | 22.3 % |
| تخم مرغ | 12.5 گرم | 0.43 گرم | 3.4 % |
| شیر گاو 3.7 درصد چربی | 3.3 گرم | 0.07 گرم | 2.1 % |
| گردو | 15 گرم | 0.82 گرم | 5.4 % |
| دانه کدو تنبل | 30 گرم | 1.85 گرم | 6.1 % |
| آرد گندم کامل | 14 گرم | 0.55 گرم | 4.0 % |
| آرد ذرت سبوس دار | 7.0 گرم | 0.28 گرم | 4.1 % |
| برنج ، پوست کنده نشده | 8.0 گرم | 0.39 گرم | 4.9 % |
| سویا ، خشک شده | 36.5 گرم | 1.9 گرم | 5.2 % |
| نخود ، خشک شده | 24.5 گرم | 1.1 گرم | 4.4 % |

همه این غذاها تقریباً منحصراً حاوی گلیسین با پیوند شیمیایی به عنوان یک جزء پروتئینی هستند، اما گلیسین آزاد ندارند.
گلایسین اولین بار در سال 2009 در نمونه های ذرات کمای دنباله دار که توسط فضاپیمای Stardust 2004 در نزدیکی 81P/Wild 2 جمع آوری شده بود، شناسایی شد . [14] [15] شناسایی شد در سال 2016، همچنین در دم دنباله دار 67P/Churyumov-Gerasimenko . [16]
توابع
متابولیسم
علاوه بر تولید گلیسین، تبدیل سرین به گلیسین برای تبدیل اسید تتراهیدروفولیک به N 5 – N 10 – متیلن – تتراهیدروفولیک اسید (TH4) که از جمله برای سنتز تیمین نوکلئوتیدهای DNA مورد نیاز است، عمل می کند. جزء ).
برعکس، با جذب CH 3 از TH4، گلیسین می تواند برای سنتز سرین استفاده شود، که سپس برای سنتز پروتئین در دسترس است، به عنوان ماده اساسی کولین یا به عنوان پیروات.
گلیسین همچنین اغلب برای سنتز سایر اجزای ماده ژنتیکی ( پورین ها ) مورد نیاز است.
همچنین بیوسنتز هِم ( اتصال به اکسیژن در خون )، کراتین (ذخیره انرژی در عضلات) یا گلوتاتیون را انجام می دهد :
- گلیسین + سوکسینیل کوآ → اسید 5 آمینولولینیک → سنتز پورفیرین برای ایجاد هم.
- گروه گلایسین + گوانیدین (از آرژنین ) → گوانیدینواستات ، که سپس می تواند وارد سنتز کراتینین شود.
- پیوند پپتیدی گلیسین + Glu-Cys → گلوتاتیون اسید
به عنوان یک محصول جانبی، اسید اگزالیک مضر نیز می تواند از گلیسین تشکیل شود.
به عنوان بخشی از متابولیسم گلیسین به عنوان یک اسید آمینه گلوکوژنیک یا گلوکوپلاستیک می تواند از طریق پیرووات به گلوکز تبدیل شود .
ماده پروتئینی
گلیسین به دلیل اندازه کوچکش ترجیحاً به پلی پپتیدها در موقعیت های محدود فضایی ( ساختار ثانویه پروتئین ) وارد می شود.
این به ویژه در کلاژن رایج ترین پروتئین موجود در موجودات حیوانی رایج است. خود بپیچد در اینجا یک سوم خوب از تمام اسیدهای آمینه را تشکیل می دهد، زیرا اندازه کوچک آن به کلاژن اجازه می دهد تا به ساختار مارپیچ سه گانه .
سیستم عصبی
گلیسین در سیستم عصبی مرکزی از طریق گیرنده گلیسین به عنوان مهاری یک انتقال دهنده عصبی ، یعنی به عنوان یک ماده سیگنال دهی بازدارنده عمل می کند. این با باز کردن کانالهای کلرید دردار لیگاند عمل میکند و منجر به پتانسیل پس سیناپسی مهاری (IPSP) میشود که فعالیت نورون پاییندستی را کاهش میدهد.
بر روی گیرنده NMDA از سوی دیگر، علاوه بر آگونیست اصلی گلوتامات ، اثر تحریکی بر روی محل اتصال ویژه گلیسین دارد.
سلولهای عصبی آزادکننده گلیسین (نرونهای گلیسینرژیک) عمدتاً در ساقه مغز و نخاع یافت میشوند [17] ، در دومی فعالیت نورونهای حرکتی شاخ قدامی را مهار میکنند و در نتیجه باعث کاهش فعالیت ماهیچههای عصبشونده میشوند. این سلول ها
که آزادسازی گلیسین را مهار می کند باعث کاهش اثر گلیسین استریکنین که به عنوان یک آنتاگونیست محل اتصال گیرنده گلیسین را مسدود می کند و سم کزاز می شود . مسدود کردن گیرنده های گلیسین یا کاهش سطح گلیسین، مهار فعالیت نورون حرکتی را کاهش می دهد که می تواند منجر به تشنج های تهدید کننده زندگی شود.
تجمع غیر طبیعی گلیسین می تواند منجر به انسفالوپاتی گلیسین شود .
استفاده
گلیسین به عنوان تقویت کننده طعم به غذاها اضافه می شود .
تایید شده است گلیسین و نمک سدیم آن به طور کلی در اتحادیه اروپا به عنوان یک افزودنی غذایی با شماره E 640 بدون حداکثر محدودیت برای غذاها و هیچ اثر منفی بر سلامتی شناخته شده ای وجود ندارد.
علاوه بر این، گلیسین جزء محلول های تزریقی برای تغذیه تزریقی است. [18]
در رزکسیون تک قطبی ترانس پیشابراه، گلیسین را می توان علاوه بر مخلوطی از مانیتول و سوربیتول به عنوان افزودنی به مایع شستشو استفاده کرد . [19]
در تحقیقات بیولوژیکی و بیوشیمیایی مولکولی، گلیسین TRIS -glycine به شکل یک سیستم بافر برای جداسازی پروتئین با استفاده از SDS-PAGE استفاده می شود. یون های گلیسین به عنوان یون های پیرو در ژل انباشته عمل می کنند. [20]